Urea Cycle Defects
Overview and Clinical Importance
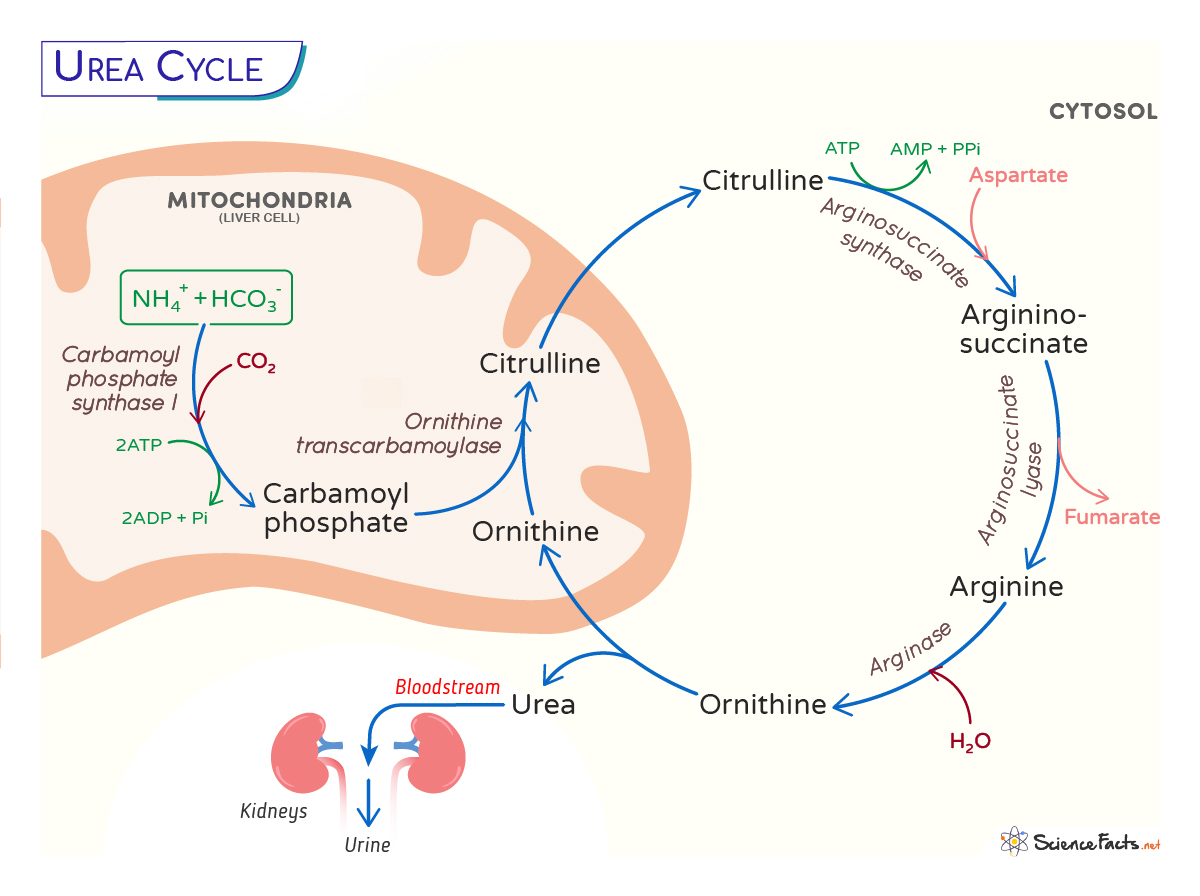
The urea cycle is the principal pathway for disposal of nitrogen derived from amino acid catabolism. It converts toxic ammonia to urea for renal excretion. Failure of the cycle causes hyperammonemia, which is neurotoxic and can produce encephalopathy, cerebral edema, developmental delay, and death if untreated.
Cellular Localization
- Mitochondrial steps: formation of carbamoyl phosphate by CPS1 and synthesis of citrulline by OTC occur in the mitochondrial matrix.
- Cytosolic steps: conversion of citrulline to argininosuccinate (ASS1), cleavage to arginine and fumarate (ASL), and hydrolysis of arginine to urea and ornithine (ARG1) occur in the cytosol.
Overall Stoichiometry
Conceptual net reaction:
NH3 + CO2 + aspartate + 3 ATP → urea + fumarate + 2 ADP +
AMP + 2 Pi
{kind=link}
Stepwise Pathway
| Step | Substrate | Enzyme (abbrev.) | Product | Compartment |
|---|---|---|---|---|
| 1 | NH3 + CO2 + 2 ATP | Carbamoyl phosphate synthetase I (CPS1) | Carbamoyl phosphate | Mitochondria |
| 2 | Carbamoyl phosphate + Ornithine | Ornithine transcarbamylase (OTC) | Citrulline | Mitochondria → Cytosol |
| 3 | Citrulline + Aspartate + ATP | Argininosuccinate synthetase (ASS1) | Argininosuccinate | Cytosol |
| 4 | Argininosuccinate | Argininosuccinate lyase (ASL) | Arginine + Fumarate | Cytosol |
| 5 | Arginine | Arginase (ARG1) | Urea + Ornithine | Cytosol |
Key Metabolic Links
- Fumarate produced by ASL enters the TCA cycle (converted to malate → oxaloacetate) and provides aspartate via transamination, linking the urea cycle and Krebs cycle.
- Ornithine is recycled into mitochondria to continue the cycle.
Regulation
- Allosteric activation: N-acetylglutamate (NAG) is an essential allosteric activator of CPS1; without adequate NAG, CPS1 activity is minimal.
- NAG synthesis: NAG synthase (NAGS) uses acetyl-CoA and glutamate; its activity is stimulated by arginine.
- Transcriptional regulation: High-protein diet, glucocorticoids, and fasting increase expression of urea cycle enzymes.
- Acute control: substrate availability (ammonia and aspartate) influences flux.
Clinical Disorders of the Urea Cycle
Inherited defects in each enzyme produce distinct biochemical patterns and clinical syndromes; most are autosomal recessive except OTC deficiency, which is X-linked.
Major enzyme deficiencies (IMAGE)
- NAGS deficiency — impaired NAG production →
CPS1 inactivity; presents like CPS1 deficiency; treatable with
carglumic acid (NAG analog).
- CPS1 deficiency — neonatal hyperammonemia; low or normal urinary orotic acid; very high ammonia, low BUN.
- OTC deficiency (X-linked) — most common urea cycle disorder; neonatal male severe hyperammonemia; increased urinary orotic acid from carbamoyl phosphate overflow into pyrimidine synthesis; heterozygous females variable.
- ASS1 deficiency (Citrullinemia type I) — elevated plasma citrulline; variable severity; risk of hyperammonemia.
- ASL deficiency (Argininosuccinic aciduria) — elevated argininosuccinate and citrulline; chronic hepatic and neuro manifestations; hypertension, trichorrhexis nodosa may be seen.
- ARG1 deficiency (Argininemia) — elevated arginine; progressive spasticity, developmental regression; less severe hyperammonemia than proximal defects.
Comparison Table of Urea Cycle Enzyme Defects
Urea Cycle Defects Comparison
| Enzyme Deficiency | Biochemical Features | Clinical Presentation | Diagnostic Clues | Treatment |
|---|---|---|---|---|
| NAGS deficiency | ↓ NAG → CPS1 inactive | Similar to CPS1 deficiency | Mimics CPS1; no orotic acid elevation | Carglumic acid (NAG analog) |
| CPS1 deficiency | ↑ Ammonia, ↓ BUN | Neonatal hyperammonemia | Low/normal urinary orotic acid | Protein restriction, ammonia scavengers |
| OTC deficiency | ↑ Ammonia, ↑ Orotic acid | Most common UCD; severe in males | X-linked; females variable; pyrimidine overflow | Protein restriction, ammonia scavengers |
| ASS1 deficiency | ↑ Citrulline | Citrullinemia type I; variable severity | Markedly elevated plasma citrulline | Arginine supplementation, ammonia control |
| ASL deficiency | ↑ Argininosuccinate, ↑ Citrulline | Chronic hepatic and neuro symptoms | Hypertension, trichorrhexis nodosa | Arginine supplementation, ammonia control |
| ARG1 deficiency | ↑ Arginine | Progressive spasticity, regression | Less severe hyperammonemia | Protein restriction; manage neurologic symptoms |
Typical Clinical Presentation
- Newborn/infant presentation: poor feeding, vomiting, decreased arousal, hypotonia, seizures, respiratory alkalosis early, progressing to somnolence, coma; often within the first 24–72 hours for severe proximal defects.
- Late/partial deficiencies: episodic lethargy, ataxia, protein intolerance, behavioral changes, developmental delay, learning disabilities, psychiatric symptoms.
- Triggers: high protein intake, illness/infection, fasting, surgery, catabolic states, steroids.
Laboratory Findings
- Markedly elevated plasma ammonia (hyperammonemia) — central finding.
- Low blood urea nitrogen (BUN).
- Respiratory alkalosis (early) from hyperventilation.
- Plasma amino acids:
- Citrulline ↑ in ASS1 deficiency
- Argininosuccinate ↑ in ASL deficiency
- Arginine ↑ in ARG1 deficiency
- Low citrulline in proximal defects (CPS1, OTC)
- Urine orotic acid:
- Elevated in OTC deficiency (carbamoyl phosphate shunted to pyrimidine synthesis).
- Normal or low in CPS1 deficiency.
- Ammonia may be >200–1000 μmol/L in neonates with severe defects (normal adult range ~11–35 μmol/L depending on lab).
Diagnostic Approach
- Measure plasma ammonia urgently in any encephalopathic infant or child.
- Obtain blood gas, glucose, electrolytes, liver panel, coagulation studies, and BUN.
- Plasma amino acid profile (citrulline, argininosuccinate, arginine, glutamine) and urine orotic acid.
- Genetic testing / targeted sequencing for suspected enzyme defect; enzyme assays from fibroblasts or liver in some cases.
- Consider newborn screening results and family history; OTC deficiency may present later in heterozygous females or with milder disease in partial deficiencies.
Acute Management of Hyperammonemic Crisis
Acute hyperammonemia is a medical emergency: reduce ammonia rapidly and protect the brain.
- Stabilize: airway, breathing, circulation. Control seizures and intracranial hypertension.
- Stop protein intake immediately (NPO) while providing nonprotein calories (glucose, lipids) to suppress catabolism.
- Provide IV fluids and high glucose infusion to reduce endogenous protein breakdown.
- Ammonia removal:
- Hemodialysis is the fastest and most effective method for rapidly lowering ammonia and is indicated for very high levels or neurologic compromise.
- Peritoneal dialysis is less effective and slower.
- Nitrogen scavenger drugs:
- Sodium benzoate: conjugates glycine to form hippurate, excreted renally.
- Sodium phenylbutyrate / phenylacetate: converted to phenylacetylglutamine, excreted renally.
- Substrate supplementation:
- Arginine (or citrulline depending on defect) to promote residual urea cycle flux and provide an alternative nitrogen acceptor; arginine is contraindicated in ARG1 deficiency.
- Carglumic acid (N-carbamylglutamate) for NAGS deficiency and some CPS1-like presentations to activate CPS1.
- Treat precipitants such as infection; correct hypoglycemia and electrolyte abnormalities.
Long-term Management
- Dietary protein restriction: carefully titrated to growth needs; specialized amino-acid–based formulas that omit problematic substrates; monitor growth, nutrition, vitamins, minerals.
- Nitrogen-scavenging drugs (oral sodium phenylbutyrate or glycerol phenylbutyrate) for chronic reduction of nitrogen load.
- Supplementation: essential amino acids as needed; citrulline or arginine depending on enzyme defect to enhance residual cycle function.
- Regular monitoring: plasma ammonia, amino acids, growth parameters, neurodevelopment, liver function.
- Liver transplantation: definitive curative option for many enzymatic defects (restores urea cycle activity). Consider for recurrent hyperammonemia despite optimal medical therapy or progressive liver disease.
- Psychosocial and developmental support: neurodevelopmental surveillance, school accommodations, genetic counseling.
Special Considerations
- OTC deficiency in females: X-inactivation causes variable presentation from mild to severe; consider in unexplained hyperammonemia across ages.
- Milder/late-onset presentations: can present in adolescence/adulthood with psychiatric symptoms, episodic encephalopathy, or protein aversion.
- Perioperative and pregnancy management: avoid catabolic states; coordinate with metabolic specialists; consider IV dextrose, continue scavenger therapy, monitor ammonia closely.
- Newborn screening: some urea cycle disorders are detected by elevated citrulline (ASS1) or other markers; however, proximal defects (CPS1, OTC) may be missed and present acutely.
Prognosis
Prognosis varies by defect severity, timing of diagnosis, and speed of treatment. Early recognition and aggressive management of hyperammonemia improve neurologic outcome. Liver transplantation offers definitive biochemical correction for many enzyme defects but carries transplant-related risks and may not reverse pre-existing neurologic injury.
Summary — Rapid Reference
| Topic | Key Points |
|---|---|
| Primary function | Convert ammonia to urea for renal excretion |
| Critical activator | N-acetylglutamate (NAG) activates CPS1; NAGS deficiency treatable with carglumic acid |
| Most urgent test | Plasma ammonia in any unexplained encephalopathy |
| Fastest ammonia removal | Hemodialysis |
| Common therapies | Protein restriction, nitrogen scavengers, arginine/citrulline supplementation, liver transplant |
| Distinct diagnostic marker | Urine orotic acid ↑ in OTC deficiency; plasma citrulline/argininosuccinate patterns identify specific defects |
https://medlineplus.gov/genetics/condition/n-acetylglutamate-synthase-deficiency/